Visium guided tutorial
Julie Bavais & Denis Puthier
2025-10-15
Source:vignettes/spatial.Rmd
spatial.RmdWorking with spatial transcriptomics datasets
The scigenex package offers a certain number of functions dedicated to spatial transcriptomic data analysis. At the moment these functions have been mainly developed to analyse VISIUM technology (10X Genomics).
Pre-process spatial transcriptomics data
Here we will use the stxBrain dataset as example. This dataset is available from SeuratData library and contains mouse brain spatial expression over in several datasets. Two datasets are for the posterior region, two for the anterior. We will use the anterior1 dataset that we will first pre-process using Seurat.
## Warning: replacing previous import 'SeuratObject::JS' by 'htmlwidgets::JS' when
## loading 'scigenex'
library(ggplot2)
library(patchwork)
suppressWarnings(SeuratData::InstallData("stxBrain"))
brain1 <- LoadData("stxBrain", type = "anterior1")
brain1 <- NormalizeData(brain1,
normalization.method = "LogNormalize",
verbose = FALSE)
brain1 <- ScaleData(brain1, verbose = FALSE)
brain1 <- FindVariableFeatures(brain1, verbose = FALSE)
brain1 <- RunPCA(brain1, assay = "Spatial", verbose = FALSE)
brain1 <- FindNeighbors(brain1, reduction = "pca", dims = 1:20, verbose = FALSE)
brain1 <- FindClusters(brain1, verbose = FALSE)
brain1 <- RunUMAP(brain1, reduction = "pca", dims = 1:20, verbose = FALSE)## Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
## To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
## This message will be shown once per session
DimPlot(brain1, reduction = "umap", label = TRUE)## Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
## ℹ Please use tidy evaluation idioms with `aes()`.
## ℹ See also `vignette("ggplot2-in-packages")` for more information.
## ℹ The deprecated feature was likely used in the Seurat package.
## Please report the issue at <https://github.com/satijalab/seurat/issues>.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.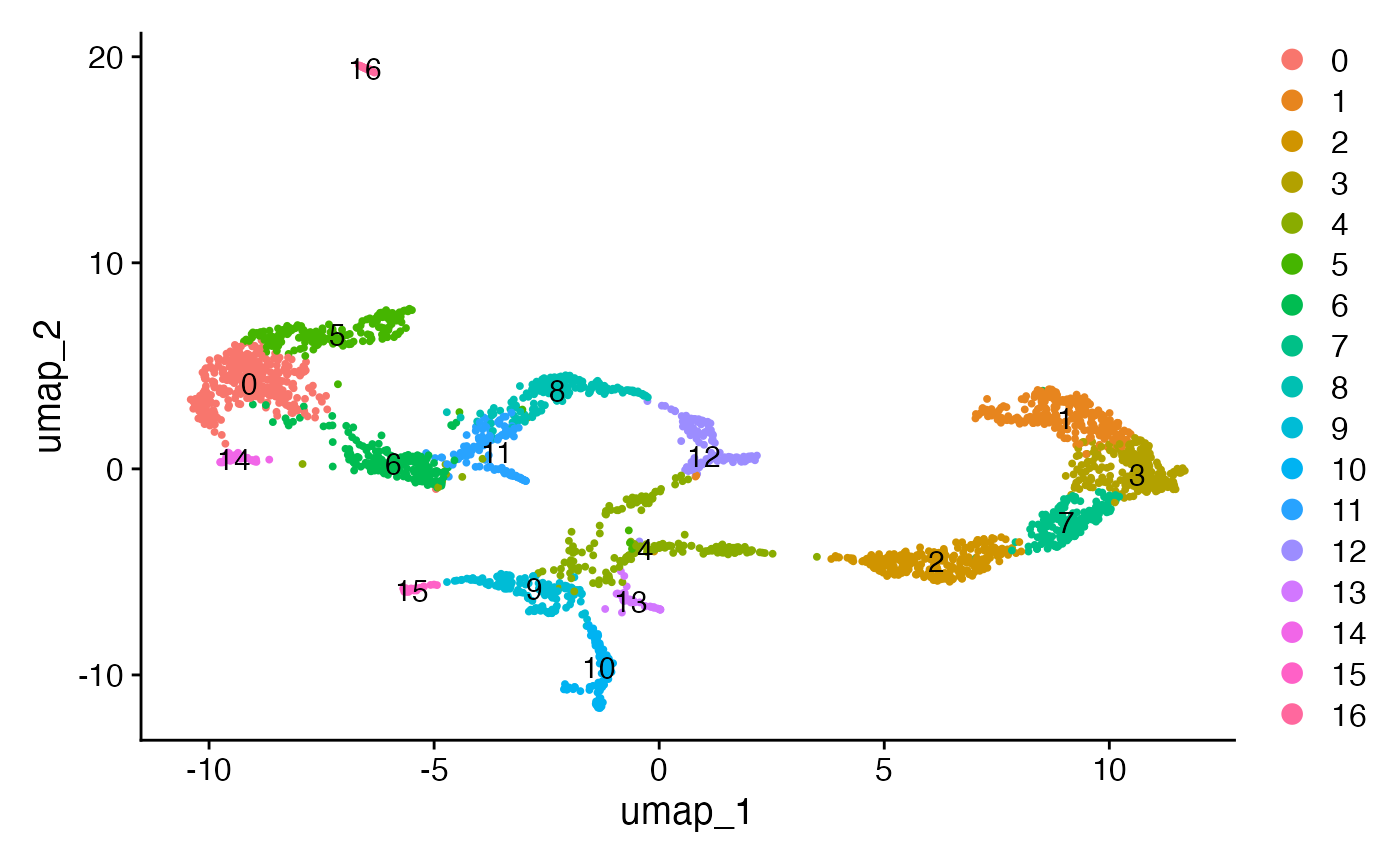
SpatialDimPlot(brain1, label = TRUE, label.size = 3, pt.size.factor = 1.4)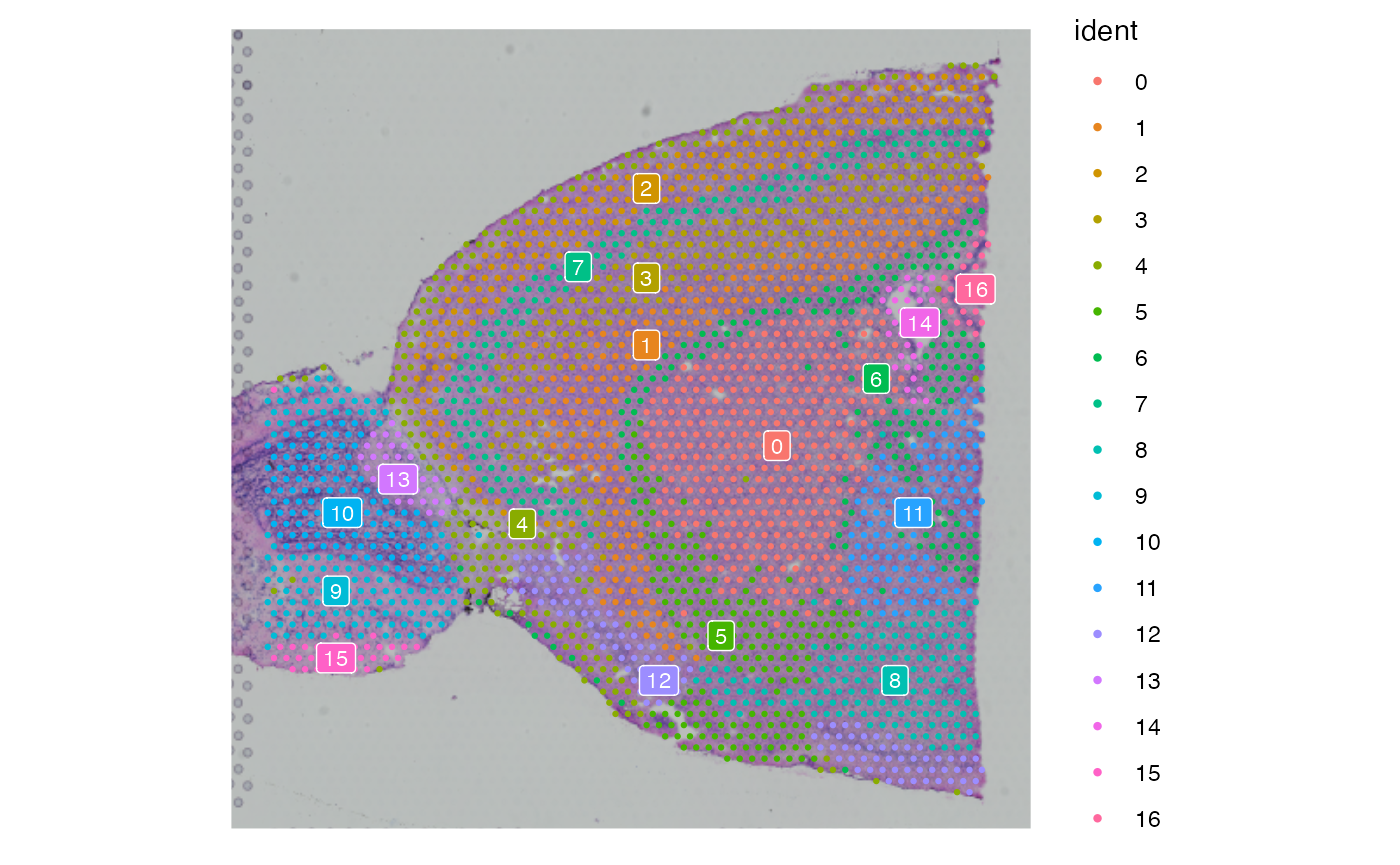
Calling scigenex
Here we will used the alternative clustering methods proposed by
scigenex (“reciprocal_neighborhood”). It is advise to increase slightly
k (here k will be set to 80). After call to
gene_clustering() we apply filtering on gene modules based
on cluster size (min number of genes 7) and standard deviation (gene
module sd > 0.1).
set_verbosity(0)
res_brain <- select_genes(data=brain1,
k=80,
distance_method="pearson",
assay="Spatial",
row_sum = 5)
gc_brain <- gene_clustering(res_brain,
method = "reciprocal_neighborhood",
inflation = 2,
threads = 4)
set_verbosity(1)
gcs_brain <- filter_cluster_size(gc_brain, min_cluster_size = 7)## |-- INFO : 53 clusters with less than 7 were filtered out.
## |-- INFO : Number of clusters left 27
df <- cluster_stats(gcs_brain)
gcss_brain <- gcs_brain[df$sd > 0.1, ]
gcss_brain <- rename_clust(gcss_brain)
length(row_names(gcss_brain))## [1] 1328
nclust(gcss_brain)## [1] 27Interestingly, Scigenex algorithm is able to retrieve
nclust(gcss_brain) gene modules. This is most probably
reminiscent of cell complexity but also of numerous molecular pathways
that are differentially activated across the organ and unanticipated
complexity.
Visualizing corresponding heatmap
We then may display the corresponding heatmap using
plot_ggheatmap().
set_verbosity(0)
gcss_brain <- top_genes(gcss_brain)
plot_ggheatmap(gcss_brain,
use_top_genes = TRUE,
ident=Idents(brain1)) + ggtitle("All clusters (top genes)") +
theme(strip.text.y = element_text(size=3))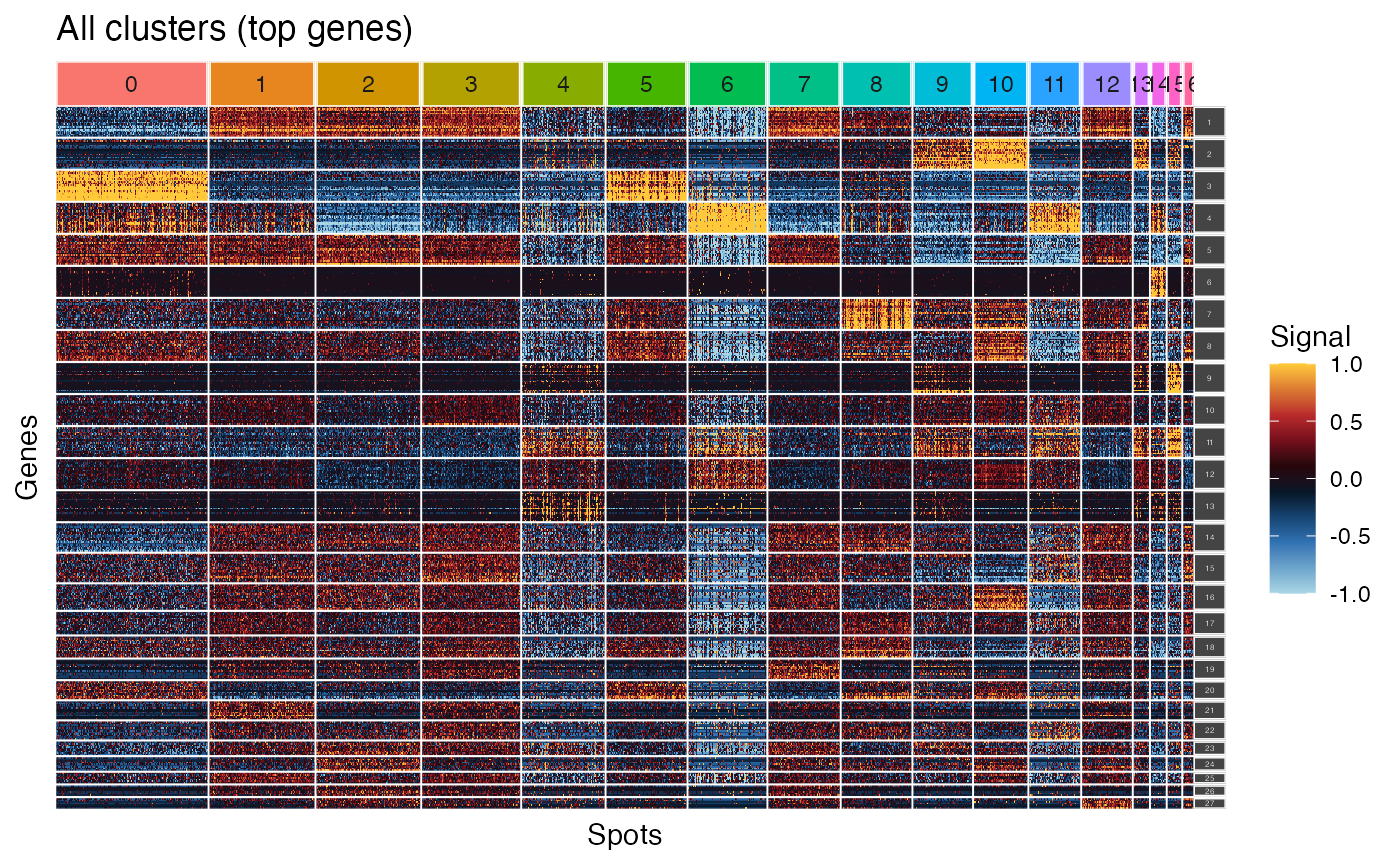
Interactive heatmap
Again, as in the context of scRNA-seq, we may also use the powerful
plot_heatmap() fonction which allows interactive
exploration of all or specific clusters. Here we look at cluster 1 to
9.
gcss_brain_sub <- subsample_by_ident(gcss_brain,
nbcell = 30,
ident = Seurat::Idents(brain1))
plot_heatmap(gcss_brain_sub[1:9,],
use_top_genes = TRUE,
cell_clusters = Seurat::Idents(brain1))