This function plots the results of compare_genesets.
plot_cmp_genesets(
set_1 = NULL,
set_2 = NULL,
stat = c("jaccard", "hypergeom", "intersection", "union", "size_set_1", "size_set_2",
"diff_set_1", "diff_set_2"),
transform = c("None", "log10", "log2", "-log10", "-log2"),
colors = colors_for_gradient("Ju1"),
layout = c("raster", "square"),
background = NULL,
coord_equal = TRUE
)Arguments
- set_1
A list containing gene sets to be compared.
- set_2
A list containing gene sets to be compared.
- stat
The statistics to be computed between gene sets. It can be either "jaccard", "hypergeom", "intersection" "size_set_1", "size_set_2", "diff_set_1" (specific to set_1), "diff_set_2" (specific to set_2). The background is taken into account. Note that hypergeometric tests check for enrichment.
- transform
The transformation to be applied to the values. It can be either "NA", "log10", or "log2", "-log10", "-log2.
- colors
The color palette to be used in the plot.
- layout
The type of diagram. Either "raster" (a scatter plot showing the statistics of interest) or "square". The "square" layout shows the hypergeometric pvalue (color) and the Jaccard result (size of the square).
- background
The background (universe) to consider. Default to the non-redundant list of elements merged from set_1 and set2. You may provide a vector with all genes of the genome for instance.
- coord_equal
make sure that the an equal length on both axis represents the same change in units
Value
A ggplot object representing the comparison results.
Details
see compare_genesets.
Examples
set.seed(123)
set_1 <- list(letters[1:10], letters[11:20], letters[21:30])
x <- sample(letters[1:30])
set_2 <- list(x[1:5], x[6:20], letters[21:30])
res <- compare_genesets(set_1, set_2, stat = "jaccard")
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 27
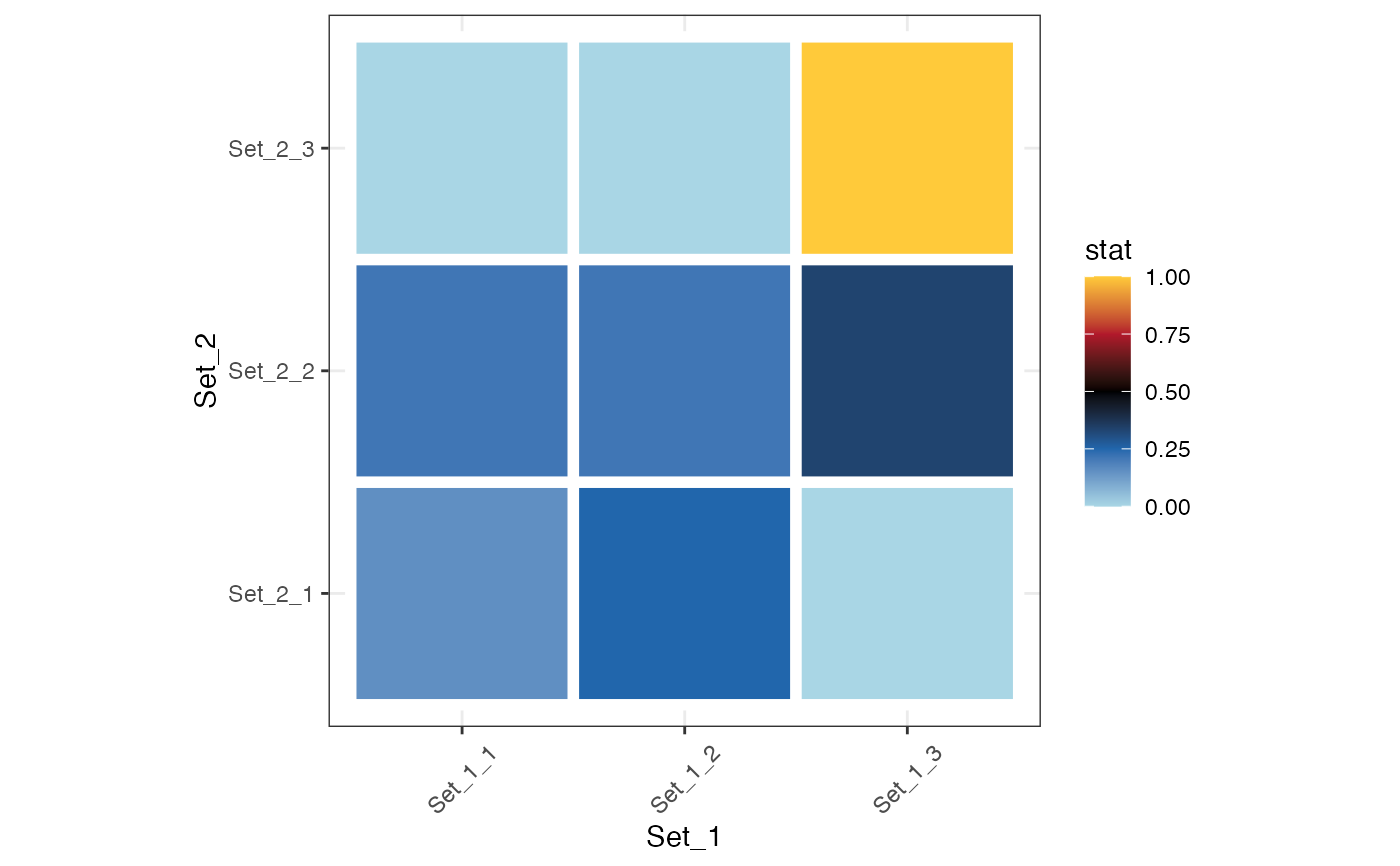
plot_cmp_genesets(set_1, set_2, stat = "jaccard")
#> |-- INFO : Using transformation: None
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 27
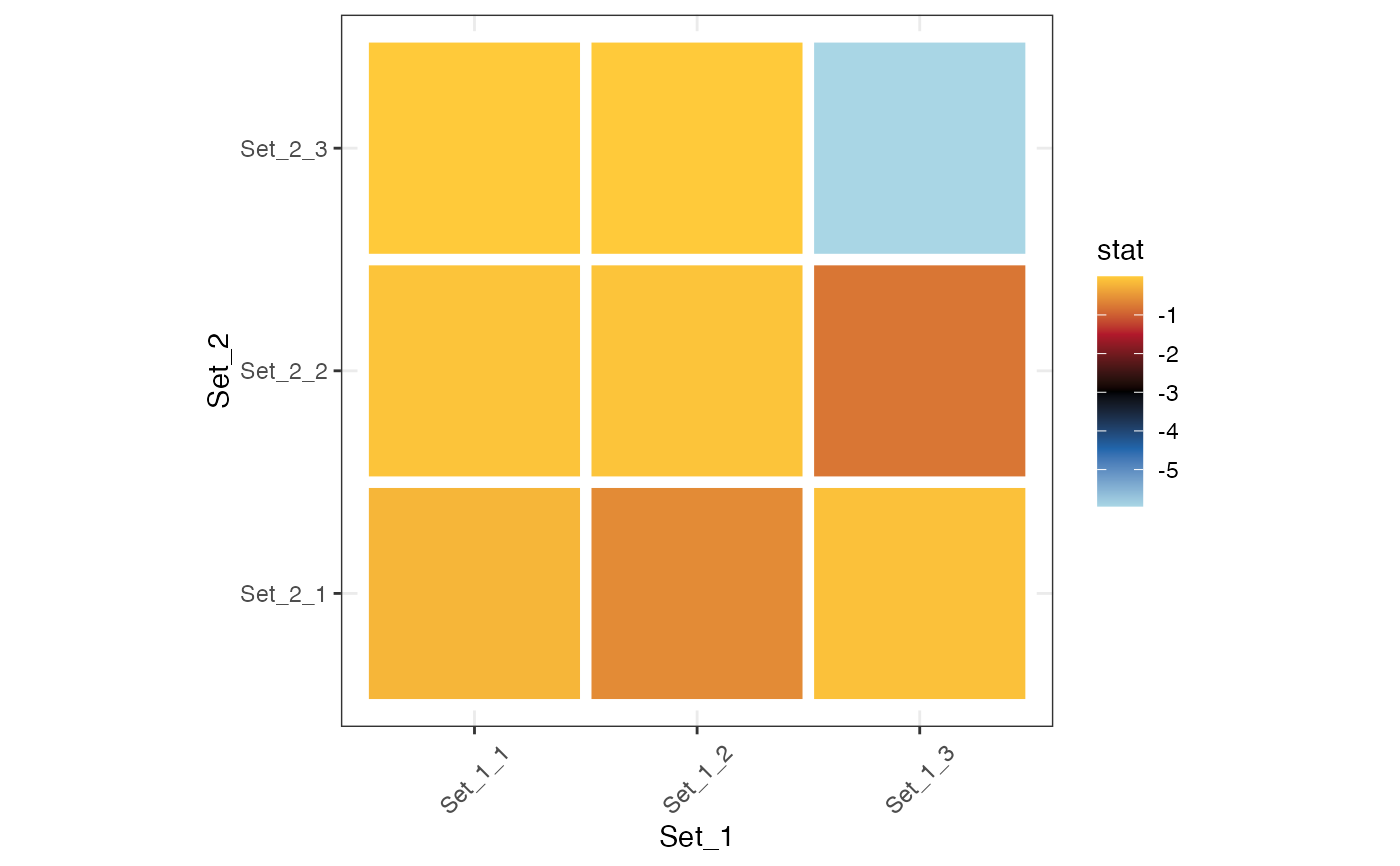
 plot_cmp_genesets(set_1, set_2, stat = "hypergeom", transform = "log10")
#> |-- INFO : Using transformation: log10
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 27
#> |-- INFO : Ceiling pvalue to 1e-320 (R limit).
plot_cmp_genesets(set_1, set_2, stat = "hypergeom", transform = "log10")
#> |-- INFO : Using transformation: log10
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 27
#> |-- INFO : Ceiling pvalue to 1e-320 (R limit).
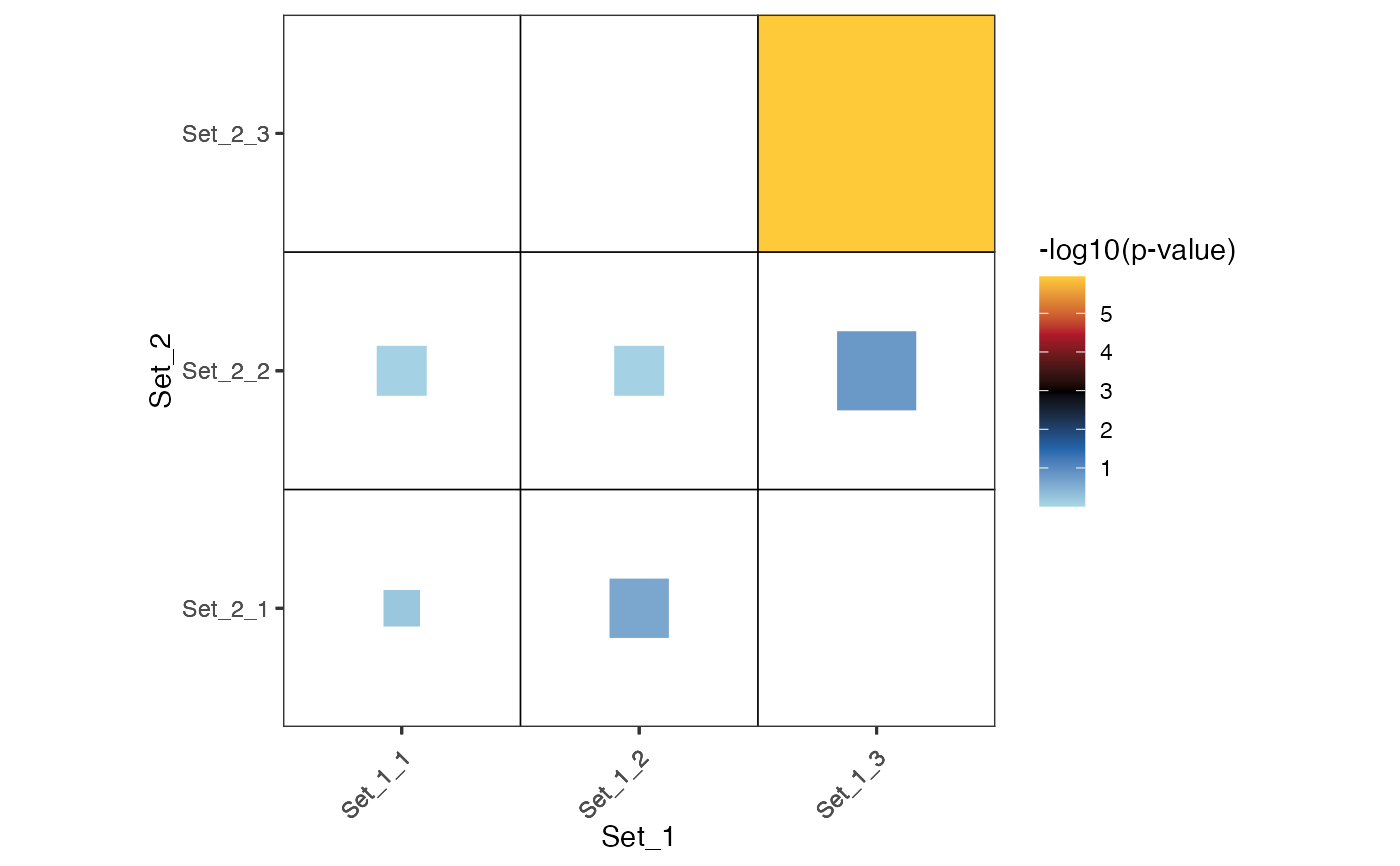
 plot_cmp_genesets(set_1, set_2, layout="square", transform = "-log10")
#> |-- INFO : Using transformation: -log10
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 27
#> |-- INFO : Ceiling pvalue to 1e-320 (R limit).
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 27
plot_cmp_genesets(set_1, set_2, layout="square", transform = "-log10")
#> |-- INFO : Using transformation: -log10
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 27
#> |-- INFO : Ceiling pvalue to 1e-320 (R limit).
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 27