Given a Seurat Spatial object, this function creates a scatter plot of the spatial expression of a gene across spots, where the X and Y coordinates represent the spatial location of spots and the color represents the expression level of the gene. This function as been tested with visium data at the moment. Defaut shape is hexagon.
plot_spatial(
seurat_obj = NULL,
gene_name = NULL,
metadata = NULL,
intensity_slot = c("data", "counts", "sct"),
title = "",
size_title = 10,
face_title = c("plain", "italic", "bold", "bold.italic"),
legend = TRUE,
barwidth = 1,
barheight = 3,
axis = TRUE,
pt_size = 3.6,
pt_shape = 6,
pt_star = TRUE,
stroke = 0,
colours = colors_for_gradient("Ju1"),
coord_flip = T
)Arguments
- seurat_obj
A Seurat object containing spatial expression data.
- gene_name
The name of the gene to plot.
- metadata
Provide the name of a metadata that will be used instead of genes (i.e. from meta.data) slot of a seurat object.
- intensity_slot
The assay slot to use for the gene expression values. Must be one of "sct", "counts", or "data". Default is "sct".
- title
The title of the plot. Default is an empty string.
- size_title
The size of the title.
- face_title
Font face for the title. Possible values are “plain”, “italic”, “bold” and “bold.italic”.
- legend
Whether to display a legend for the color scale. Default is FALSE.
- barwidth
A numeric or a grid::unit() object specifying the width of the colourbar. Default value is legend.key.width or legend.key.size in theme() or theme.
- barheight
A numeric or a grid::unit() object specifying the height of the colourbar. Default value is legend.key.height or legend.key.size in theme() or theme.
- axis
Whether to display a axis for the color scale. Default is FALSE.
- pt_size
The size of the points in the plot. Default is 2.1.
- pt_shape
The shape of the points in the plot. Default is 16 (a circle).
- pt_star
A boolean. Whether to use ggstar shapes.
- stroke
The thickness of margin of points.
- colours
A vector of colors.
- coord_flip
Whether to flip coordinates.
Value
A ggplot2 object containing the scatter plot.
Examples
library(Seurat)
load_example_dataset("7870305/files/lymph_node_tiny_2")
#> |-- INFO : Dataset 7870305/files/lymph_node_tiny_2 was already loaded.
plot_spatial(seurat_obj = lymph_node_tiny_2, gene_name = "CCL22", intensity_slot="data", pt_size=6)
#> |-- INFO : Feature is not a factor.
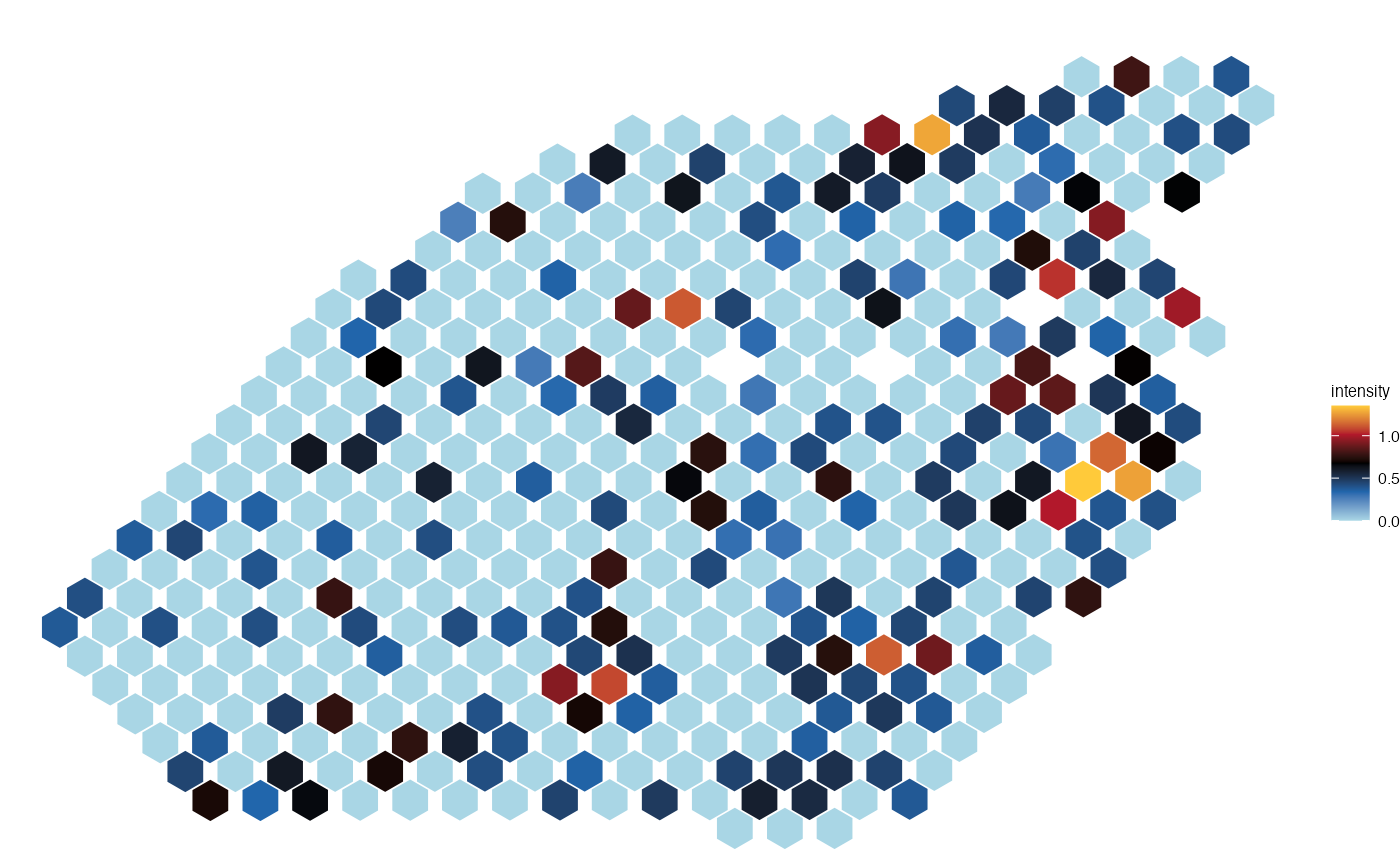 plot_spatial(seurat_obj = lymph_node_tiny_2, metadata = "nCount_Spatial", pt_size=6)
#> |-- INFO : Feature is not a factor.
plot_spatial(seurat_obj = lymph_node_tiny_2, metadata = "nCount_Spatial", pt_size=6)
#> |-- INFO : Feature is not a factor.
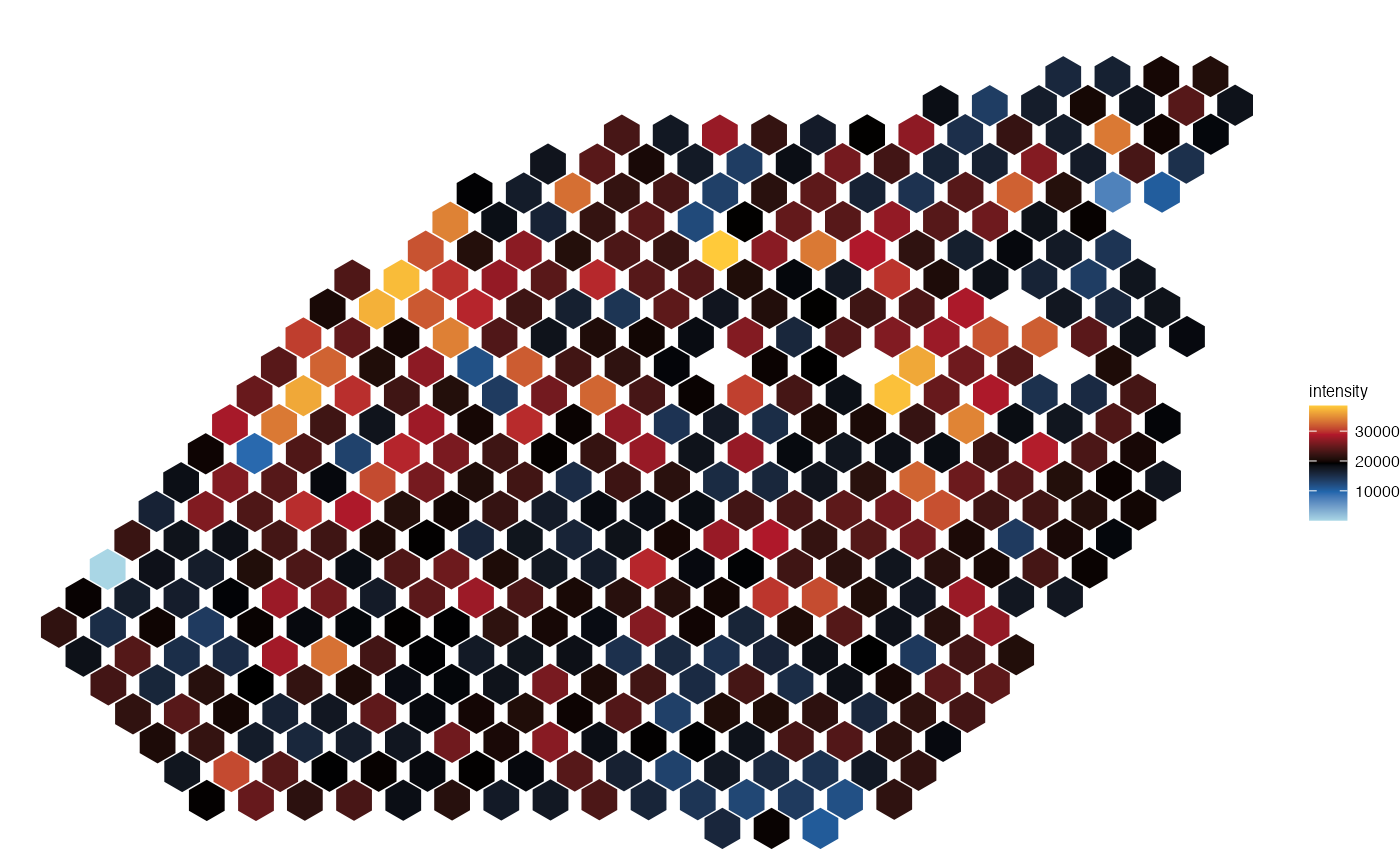 hull <- display_hull(lymph_node_tiny_2,
ident=ifelse(Seurat::Idents(lymph_node_tiny_2) %in% 7, 1, 0),
delta=1, size_x=3.4, size_y=3, color="black")
#> |-- INFO : Creating a dataframe to store output
#> |-- INFO : Creating a list of neighborhoods.
#> |-- INFO : Looping over the points.
p <- plot_spatial(seurat_obj = lymph_node_tiny_2, gene_name = "VPREB3", intensity_slot="data", pt_size=6)
#> |-- INFO : Feature is not a factor.
p + hull
hull <- display_hull(lymph_node_tiny_2,
ident=ifelse(Seurat::Idents(lymph_node_tiny_2) %in% 7, 1, 0),
delta=1, size_x=3.4, size_y=3, color="black")
#> |-- INFO : Creating a dataframe to store output
#> |-- INFO : Creating a list of neighborhoods.
#> |-- INFO : Looping over the points.
p <- plot_spatial(seurat_obj = lymph_node_tiny_2, gene_name = "VPREB3", intensity_slot="data", pt_size=6)
#> |-- INFO : Feature is not a factor.
p + hull
