A planel of XY scatter plots of Visium data with hexagonal spots.
Source:R/plot_spatial.R
plot_spatial_panel.RdThis function creates a panel of scatter plots for the spatial expression of a list of genes across spots, where the X and Y coordinates represent the spatial location of spots and the color represents the expression level of the gene.
plot_spatial_panel(
seurat_obj = NULL,
genes = NULL,
metadata = NULL,
intensity_slot = c("data", "counts", "sct"),
title = "",
size_title = 10,
face_title = c("plain", "italic", "bold", "bold.italic"),
barwidth = 1,
barheight = 3,
axis = TRUE,
panel_names = NULL,
ncol_layout = NULL,
legend = TRUE,
guides = NULL,
pt_size = 3.6,
pt_shape = 6,
pt_star = TRUE,
stroke = 0,
coord_flip = TRUE,
colours = colors_for_gradient("Ju1")
)Arguments
- seurat_obj
A Seurat object containing spatial expression data.
- genes
A vector of gene names to plot.
- metadata
Provide a vector of metadata that will be used instead of genes (i.e. from meta.data) slot of a seurat object.
- intensity_slot
The assay slot to use for the gene expression values. Must be one of "sct", "counts", or "data". Default is "sct".
- title
The title of the plot. Default is an empty string.
- size_title
The size of the titles.
- face_title
Font face for the title. Possible values are “plain”, “italic”, “bold” and “bold.italic”.
- barwidth
A numeric or a grid::unit() object specifying the width of the colourbar. Default value is legend.key.width or legend.key.size in theme() or theme.
- barheight
A numeric or a grid::unit() object specifying the height of the colourbar. Default value is legend.key.height or legend.key.size in theme() or theme.
- axis
Whether to display a axis for the color scale. Default is FALSE.
- panel_names
A vector of panel names to use for each gene plot.
- ncol_layout
Number of columns to use for the panel layout. Default is the ceiling of the number of genes divided by 2.
- legend
Whether to display a legend for the color scale. Default is FALSE.
- guides
A string specifying how guides should be treated in the layout. See patchwork::plot_layout().
- pt_size
The size of the points in the plot. Default is 2.1.
- pt_shape
The shape of the points in the plot. Default is 16 (a circle).
- pt_star
A boolean. Whether to use ggstar shapes.
- stroke
The thickness of margin of points.
- coord_flip
Whether to flip coordinates.
- colours
A vector of colors.
Value
A ggplot2 object containing the panel of scatter plots.
Examples
library(Seurat)
load_example_dataset("7870305/files/lymph_node_tiny_2")
#> |-- INFO : Dataset 7870305/files/lymph_node_tiny_2 was already loaded.
load_example_dataset("7870305/files/lymph_node_tiny_clusters_2")
#> |-- INFO : Dataset 7870305/files/lymph_node_tiny_clusters_2 was already loaded.
lymph_node_tiny_2 <- Seurat::AddModuleScore(lymph_node_tiny_2, features = lymph_node_tiny_clusters_2@gene_clusters, nbin = 15)
for(i in 1:nclust(lymph_node_tiny_clusters_2)){ # Normalizing module scores
tmp <- lymph_node_tiny_2[[paste0("Cluster", i, sep="")]]
max_tmp <- max(tmp)
min_tmp <- min(tmp)
lymph_node_tiny_2[[paste0("Cluster", i, sep="")]] <- (tmp[,1] - min(tmp))/(max_tmp - min_tmp)
}
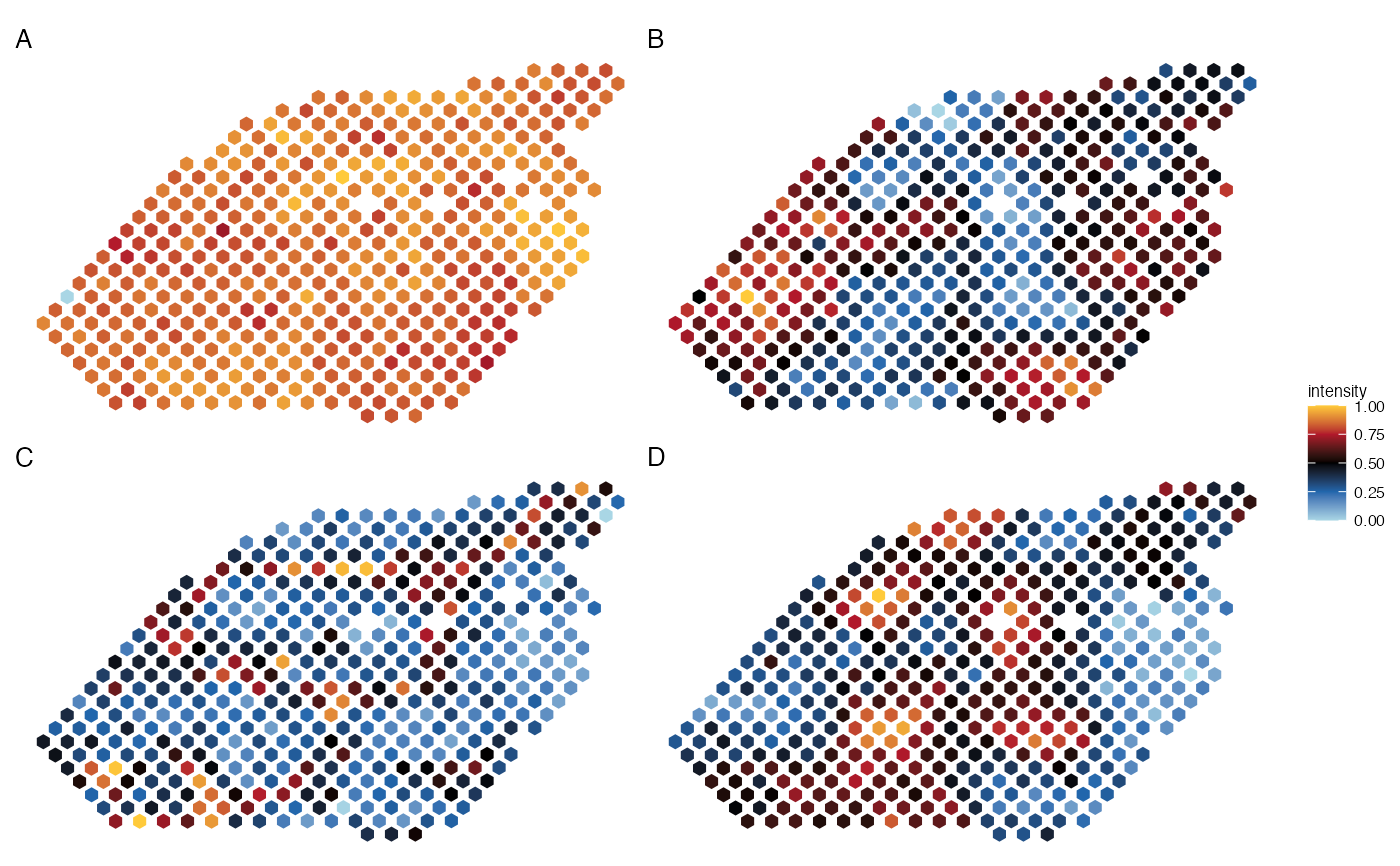
plot_spatial_panel(lymph_node_tiny_2, metadata=paste0("Cluster", 1:4), ncol_layout=2,
guides='collect', pt_size=2.2, coord_flip=TRUE)
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
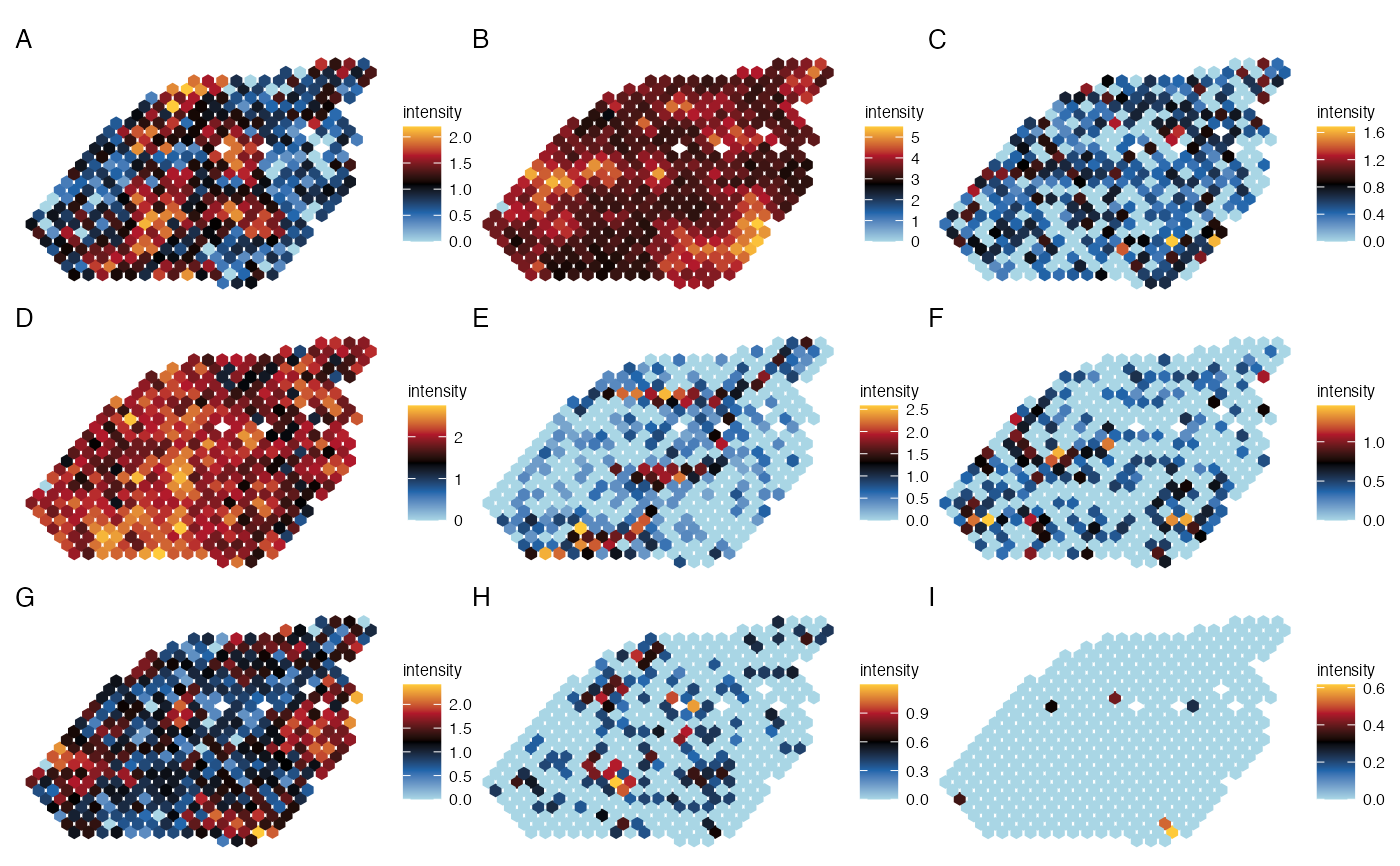
 plot_spatial_panel(lymph_node_tiny_2, gene=c('VPREB3', 'IGHG1', 'PRDX4',
'LTB', 'CCL20', 'LYVE1',
'IL7R', 'RGS9', 'MAPT'),
ncol_layout=3,
pt_size=2, coord_flip=TRUE,
panel_names=LETTERS[1:9],
size_title = 10)
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
plot_spatial_panel(lymph_node_tiny_2, gene=c('VPREB3', 'IGHG1', 'PRDX4',
'LTB', 'CCL20', 'LYVE1',
'IL7R', 'RGS9', 'MAPT'),
ncol_layout=3,
pt_size=2, coord_flip=TRUE,
panel_names=LETTERS[1:9],
size_title = 10)
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.
#> |-- INFO : Feature is not a factor.