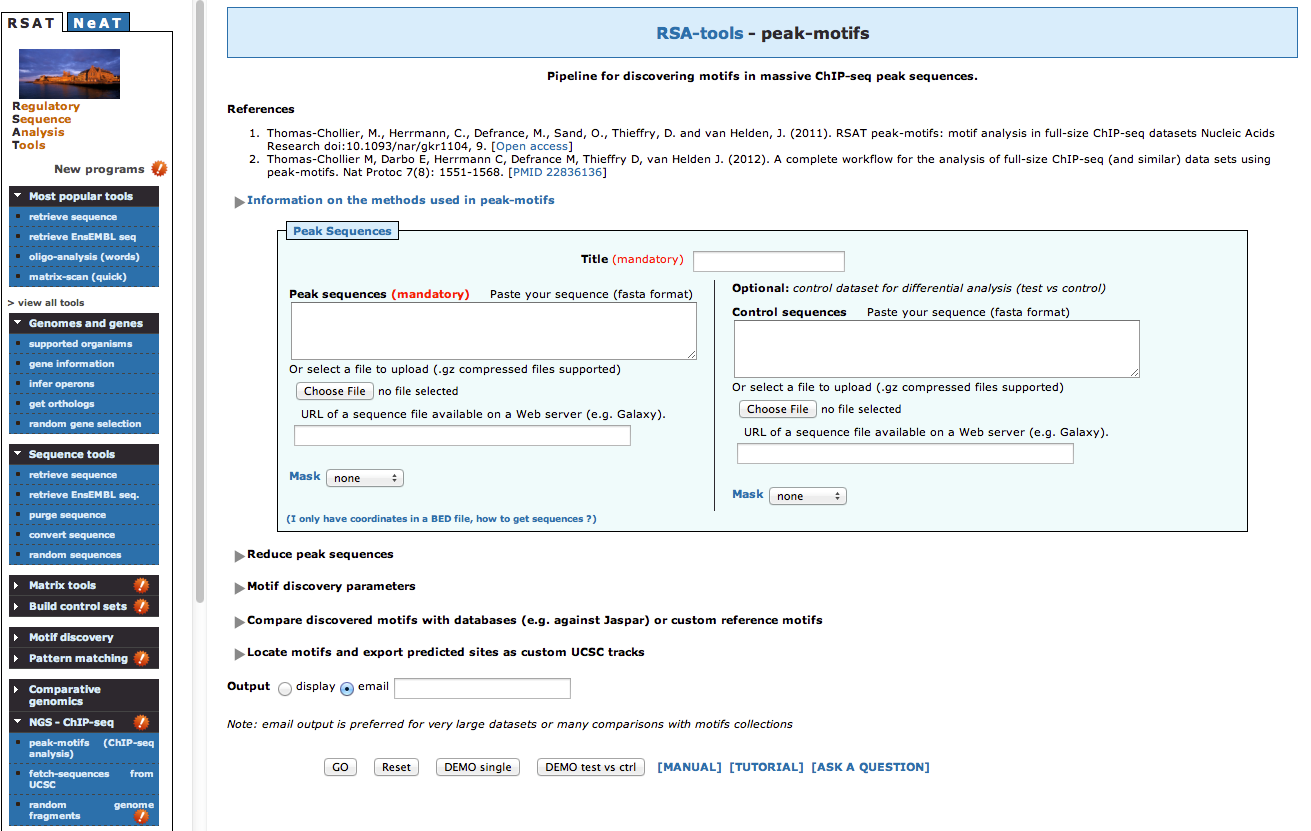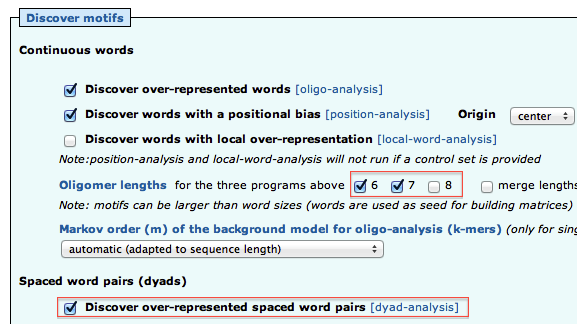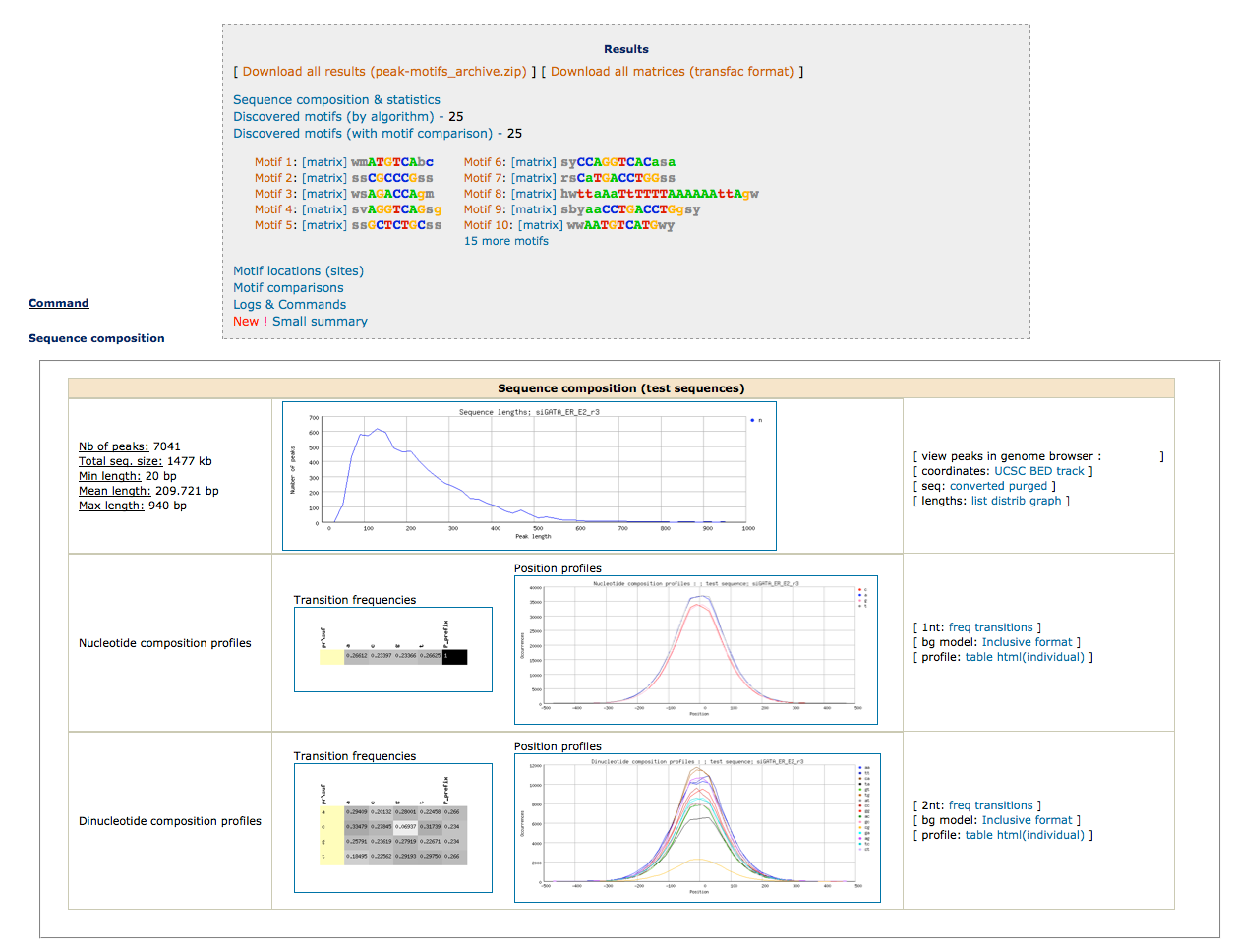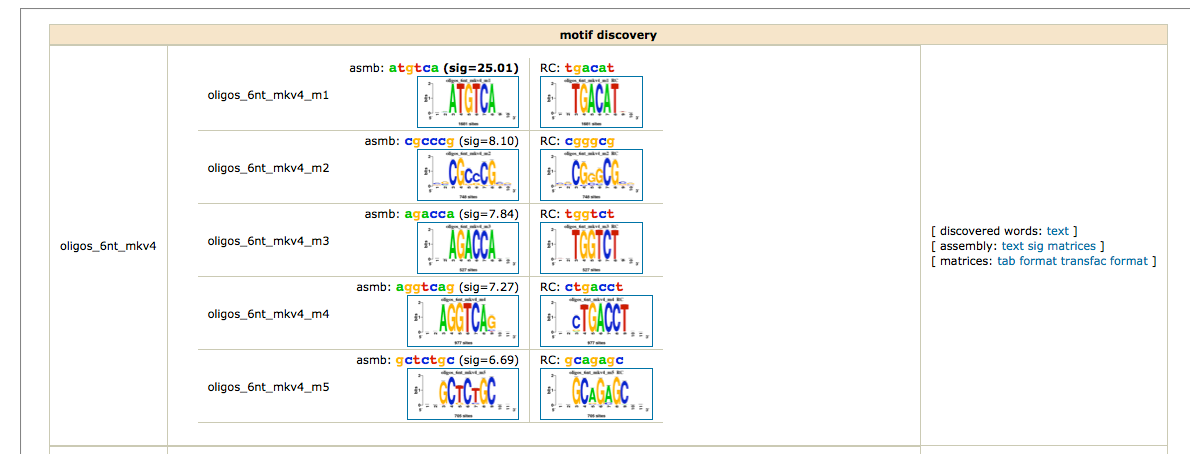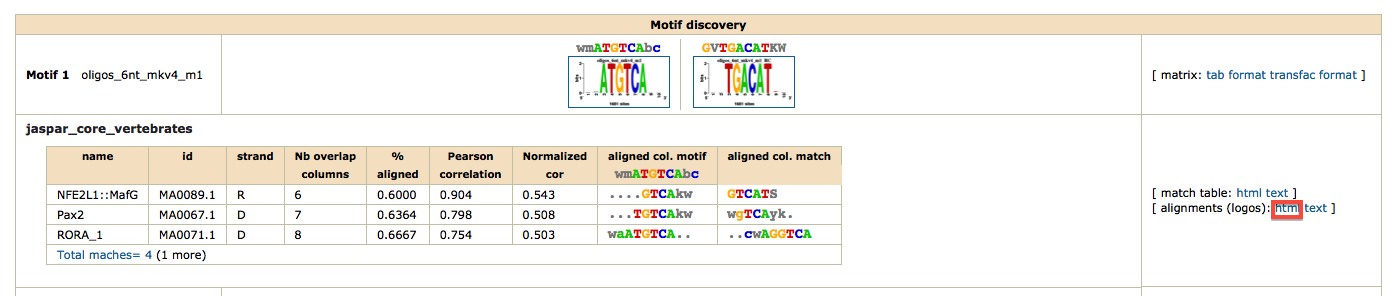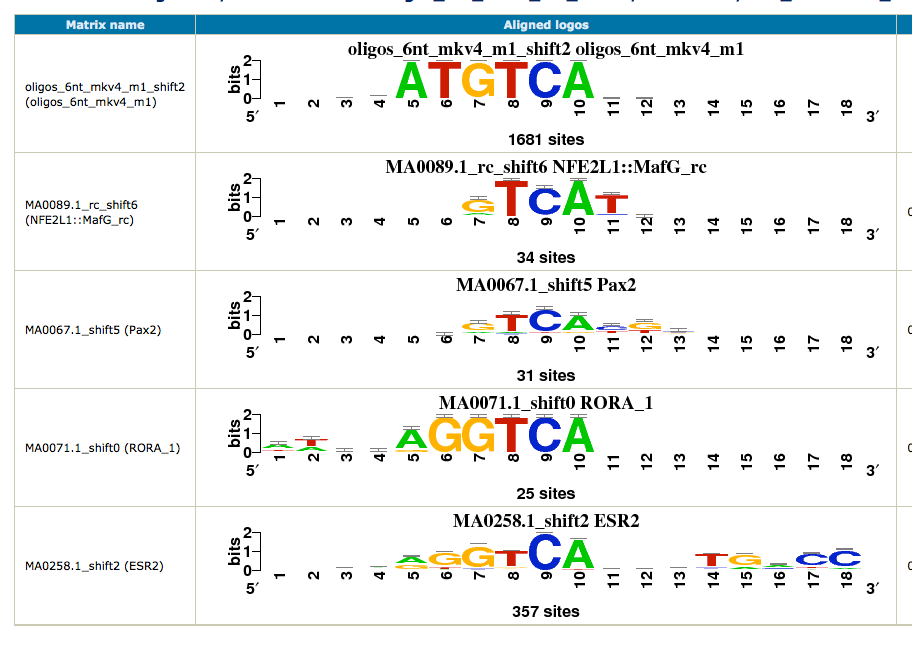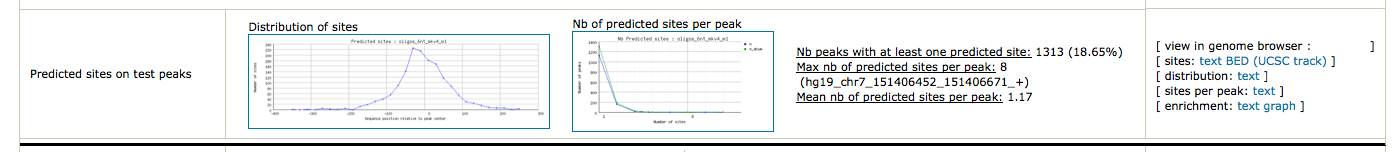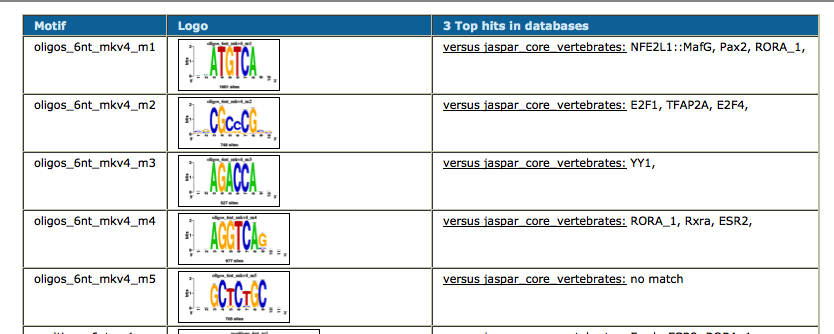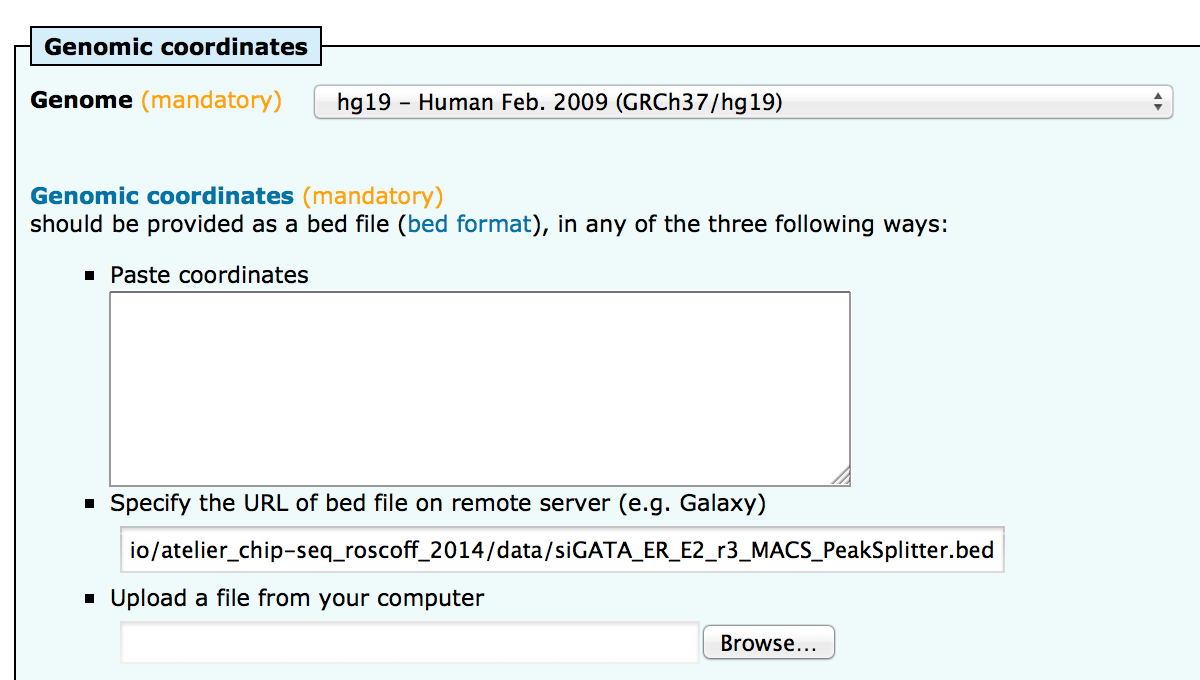
What is the result of this fetch-sequences tool ?

The program returns 3 links to files :
- the input BED file (coordinates),
- the corresponding sequences (FASTA file), and
- a log text file providing information on the execution of the program.