Map cell markers to clusters (hypergeometric/jaccard)
Source:R/enrichment_analysis.R
plot_markers_to_clusters.RdUse jaccard index and hypergeometric to map a list of cell markers to gene clusters.
plot_markers_to_clusters(
object,
markers = NULL,
gradient_palette = colors_for_gradient("Magma"),
background = NULL,
cell_type_order = c("None", "hclust"),
only_specific = FALSE,
jaccard_scale = 10
)Arguments
- object
A ClusterSet object.
- markers
A list of cell markers.
- gradient_palette
A vector of colors for the gradient palette.
- background
The background for hypergeometric test. Typically the list of genes annotated in the considered gene ontology (e.g. BP).
- cell_type_order
The order of cell types. Can be "None" or "hclust". Not supported at the moment.
- only_specific
Logical value indicating whether to show only specific markers.
- jaccard_scale
The scale factor for Jaccard coefficient.
Value
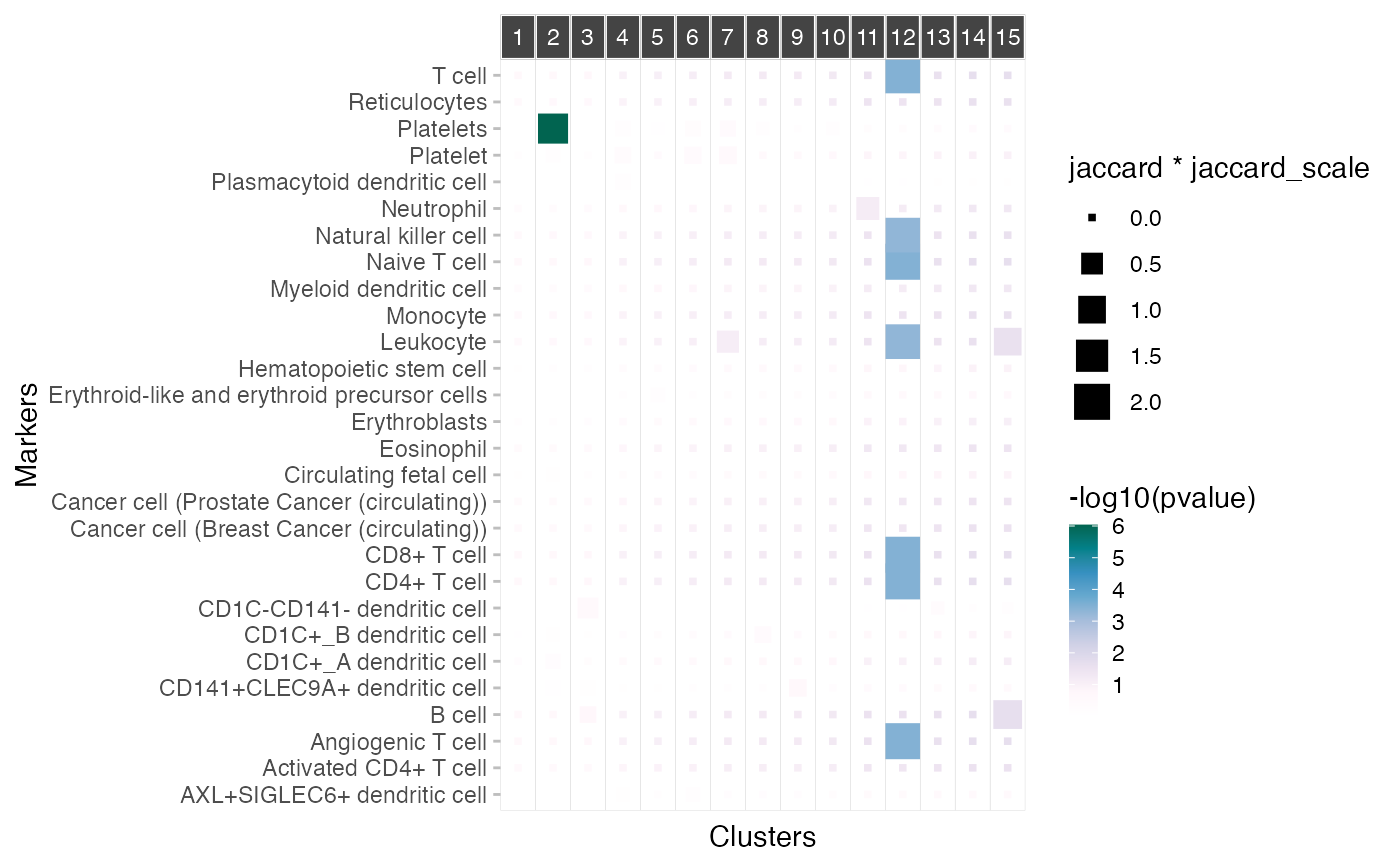
A plot showing the relationship between markers and clusters.
Examples
load_example_dataset("8028126/files/pbmc3k_medium_clusters_enr")
#> |-- INFO : Dataset 8028126/files/pbmc3k_medium_clusters_enr was already loaded.
library(clustermole)
m <- clustermole::clustermole_markers(species = "hs")
markers <- split(m$gene[m$organ == "Blood" & m$species == "Human"],
m$celltype[m$organ == "Blood" & m$species == "Human"])
plot_markers_to_clusters(pbmc3k_medium_clusters_enr, markers=markers)
#> |-- INFO : WARNING: Running without a background but you should probably provide one.
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 1096
#> |-- INFO : Computing background from the union of set_1 and set_2.
#> |-- INFO : Background size : 1096