The installation script can be downloaded here (right-click on the link and select "Copy link adress" to get the URL.).
- Go to the /tmp directory (cd).
- Using the wget command, download the installation script.
- Use the ls command with -l arguments to check that the file is present. Have a look at access permissions.
- check the content of installBedtools.sh script with less.
- Use the chmod command to give yourself (the User) eXecute permission on file installBedtools.sh.
- Using the ls command with -l arguments check access permissions.
- Execute the installBedtools.sh script.
- Instruct the terminal to reload the ~/.bashrc file (source)
- Go back to the ~/TD02_Bioinfo directory (cd).
- Type bedtools -h
Which fraction of the human genome is covered by exons ?
In the section below we will try to answer the following question: "Which fraction of the genome is covered by exons ?".
Downloading genomic coordinates of exons.
First, we will retrieve feature coordinates (exons) from the UCSC server.
- Using your Web browser, go to the UCSC web site.
- Select Tables in the top menu.
- Select the following parameters:
- Clade: Mammal,
- Genome: Human,
- assembly: "Dec. 2013 (hg38, GRCh38)",
- group: Genes and Gene Prediction tracks,
- track: RefSeq Genes,
- table: refGenes,
- region: genome,
- Output format: BED.
- Set output file to "RefGene_hg38_exons.bed" and click on Get output.
- In the next window, select Exons (plus 0
bases at each end). Leave all other options
unchanged, click the button get
BED.
- Note that with the same protocol we could
also select the coordinates of all transcripts or
intronic regions).
- Use the mv command to move the file RefGene_hg38_exons.bed from ~/Téléchargements (or ~/Downloads) to the your result directory TD02_Bioinfo.
- Look at the 6 first lines of the file. Does it look like a bed format ?
- Is it a tabulated file ?
Merging overlapping regions
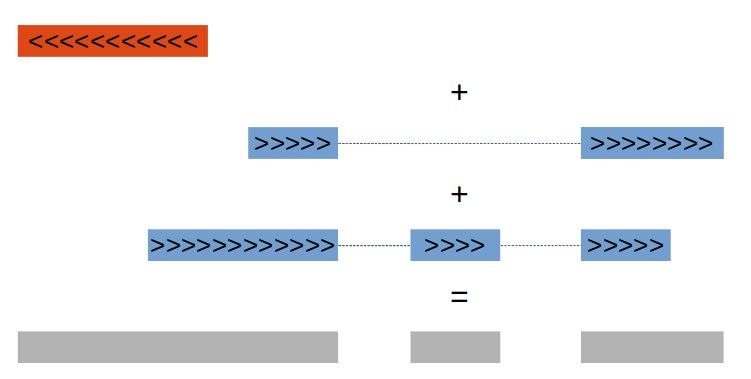
To answer our simple question, we must first keep in mind that several exons may overlap, due to various phenomena (alternative splicing, multiple promoters or terminators, mutually overlapping genes). To avoid counting several times the same region, our first task will thus be to merge these overlapping regions. The mergeBed command from the Bedtools suite combines overlapping features in an interval file into a single feature which spans all of the combined features. The image below illustrate this.
Beware: MergeBed requires the genomic coordinates to be sorted (see below).
We will first discard genes located on "non-regular" chromosomes. For this, we consider as "regular" the chromosome names starging with "chr" followed by one or more numbers (chr1, chr17,...) or the specific letters "X" or "Y" (chrX, chrY). We will first select the features from the bed files that match these regular names, and then count among them (i) the total number of exons and (ii) the number of exons per chromosome.
grep -P "^chr[0-9XY]+\t" RefGene_hg38_exons.bed > RefGene_hg38_exons_reg.bed # delete non 'regular' chromosomes.
wc -l RefGene_hg38_exons_reg.bed # Total number of exons
cut -f1 RefGene_hg38_exons_reg.bed | sort | uniq -c | sort -rn # Check the number of exon per chromosome
- Get some help about the sortBed command using the -h (help) argument.
- Use the sortBed command to sort exons by coordinates and store the results in RefGene_hg38_exons_reg_sort.bed.
- Get some help about the mergeBed command using the -h (help) argument.
- Use mergeBed with RefGene_hg38_exons_reg_sort.bed as input to combine overlapping exons into single features and store the results into a file named mergedExons.bed.
- Have a look at the first lines with the head command.
- Count the number of lines in all *.bed files using wc.. Is the result as expected?
- The length of one genomic feature can simply be obtained by computing column_3-column_2. Use a awk command to compute the sum of the length of all features.
- Compute the total length of the genome using the file ~/TD01_Bioinfo/hg38_transcripts/chromInfo.txt (see TD01). As an alternative, download the file here using wget.
- Now, what is the fraction of the genome that is covered by exons or genes ?
Genomic locations of SNPs associated with prostate cancer
Genome-Wide Association Studies (GWAS) are used in epidemiology to search for common genetics variants associated with a given disease. GWAS typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like diseases. Prostate cancer (PrCa) is the most frequently diagnosed cancer in males in developed countries. To identify common PrCa susceptibility alleles, Eeles RA et al, conducted a GWAS whose results are available through GWAS Central (study HGVST512). The top 50 associations (dataset: HGVRS986) were retrieved from GWAS Central and converted to a BED format (build hg38).
Which of these SNPs fall into exonic regions ?
The intersectBed program can be used to answer such questions (click for more informations).- Get some help about the intersectBed program (argument-h).
- Download SNPs list in BED format here.
- Use the intersectBed command with -a, -b, -wa and -wb arguments to find SNPs falling into exonic regions.
Which of these SNPs fall into intronic regions ?
- Download intronic regions as bed format here.
- Use the intersectBed command with -a, -b, -wa and -wb arguments to find SNPs falling into intronic regions.
Which of these SNPs fall into promoter regions ?
As you can see, lots of these SNPs are located in intergenic regions (i.e. outside known genes). One additional question could be whether some of them are falling into promoter regions. As the promoter regions is difficult to define without additional informations (e.g. epigenetic marks) we will define it, here as the regions ransging from the the transcriptional start site (TSS) to -500bp upstream of the TSS. To answer this question, we need to extract those regions.
- Download the coordinates of the whole transcripts here (note that you can get it also from the table browser).
- Use the following awk onliner to extract promoter region coordiantes:
wget http://pedagogix-tagc.univ-mrs.fr/courses/jgb53d-bd-prog/data/RefGene_hg38_wg_reg.bed.gz
gunzip RefGene_hg38_wg_reg.bed.gz
cut -f6 RefGene_hg38_wg_reg.bed | sort | uniq -c # ensure that all transcript strands are defined
awk 'BEGIN{FS=OFS="\t"}{if($5=="+"){print $1,$2-500,$2,$4,$5,$6}else{print $1,$3,$3+500,$4,$5,$6}}' RefGene_hg38_wg_reg.bed > RefGene_hg38_prom_reg.bed # get the promoter regions.
One of the remaining problem is that or promoter regions from transcript t may overlap with exonic regions from another transcript. We can not strictly declare them as regulatory regions. We thus can discard these overlapping regions. This can be done with the subtractBed (click for more informations) command from the Bedtools suite.
- Use the subtractBed to delete any promoter region overlapping exons (create a file named RefGene_hg38_prom_reg_noExons.bed).
- To ensure that this step was effective go to UCSC.
- From the top Menu select Genomes
- Select group: Mammal, genome: Human, assembly: "Dec. 2013 (hg38, GRCh38)".
- Click on manage custom tracks > add custom track > Choose File (browse to RefGene_hg38_prom_reg_noExons.bed) and click Submit.
- click on go to genome browser. Enter position chr1:201504430-201512040 (for instance) to check the result.
- Use intersectBed to find SNPs overlapping promoter regions.
- Get information relative to NM_138634 and its associated gene here. Is there any link with PrCa?