In this practical, we will inspect the statistical tests used to compare a set of genes of interest to a set of reference genes. This practical is essentially a tutorial, based on the result returned by David in the previous practical Handling genomic coordinates.
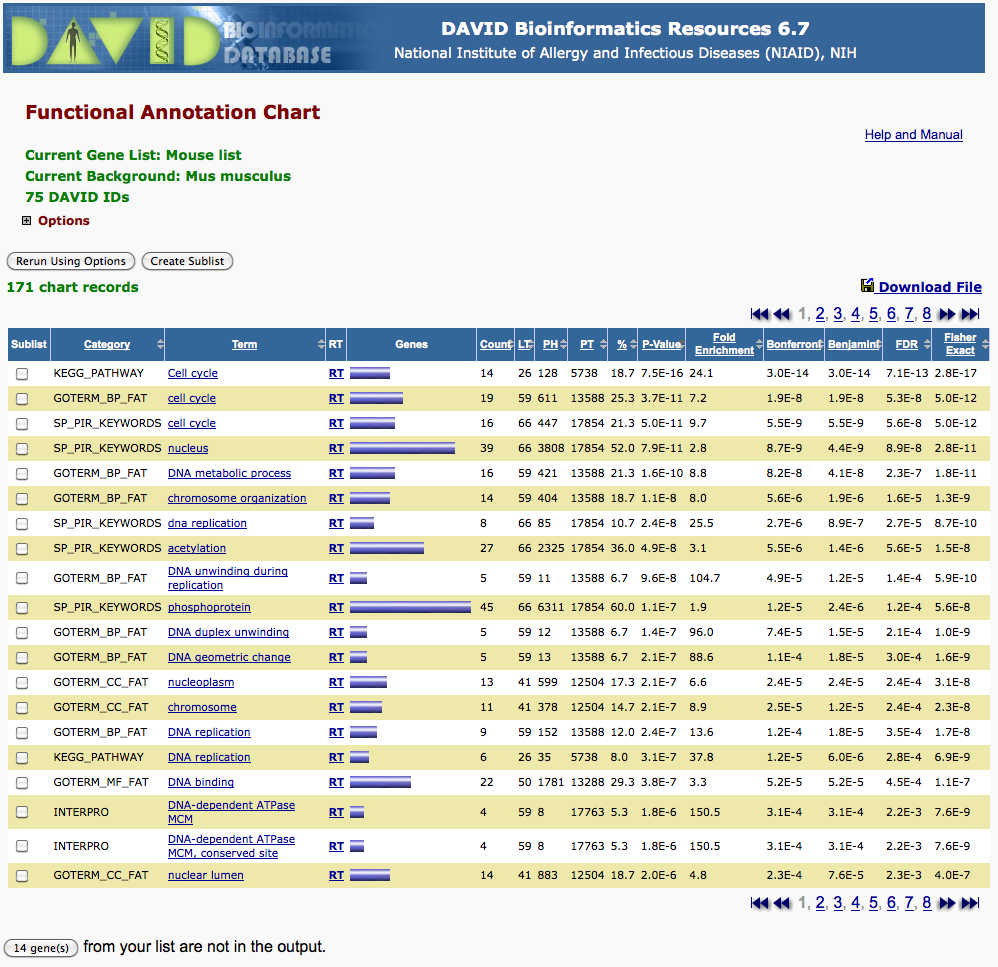
In this tutorial, we hade submitted a set of predicted E2F target genes (see file M00920_targets.txt) to the Web tool DAVID, to compare it to various catalogues of functional annotations (Gene Ontology, KEGG, ...). DAVID returned a table reporting the functional classes for which our gene set showed significant enrichment.
The goal of this tutorial is to reproduce the calculation of the significance. We will show two distinct ways to model the problem:
Click to open the image in a separate window
In a first time, we model the association between genes and GO class using a hypergeometric distribution. The classical example for the hypergeometric is the ranomd selection of "k" balls in an urn containing "m" marked and "n" non-marked balls, and the observation that the selection contains "x" marked balls.
To illustrate the way to perform the hypergeometric test, we will re-analyze the second row of the David result displayed above: correspondence between a set of predicted E2F target genes and the genes annotated in the functional class "cell cycle" of the GOTERM_BP_FAT terms.
We define the parameters of the hypergeometric test in the following way:
| m = 611 | number of "marked" elements, i.e. total number of genes annotated for the selected GO term (cell cycle in GOTERM_BP_FAT annotations). |
| N = 13588 | total number of genes with some annotation in GOTERM_BP_FAT. Note: this is lower than the total number of human genes, since many genes are of totally unknown function. |
| n = N - m | number of "non-marked" elements, i.e. the number of genes that have some annotation in GOERM_BP_FAT, but are not associated to the selected GO term (cell cycle). |
| k = 59 | Size of the selection, i.e. number of genes predicted as E2F targets, and associated to at least one "Biological Process" in the Gene Ontology. |
| x = 19 | number of "marked" elements in the selection, i.e. number of genes predicted as E2F targets AND associated to the process "cell cycle" in GO annotations. |
g <- 75 ## Number of submitted genes
k <- 59 ## Size of the selection, i.e. submitted genes with at least one annotation in GO biological processes
m <- 611 ## Number of "marked" elements, i.e. genes associated to this biological process
N <- 13588 ## Total number of genes with some annotation in GOTERM_BP_FAT.
n <- N - m ## Number of "non-marked" elements, i.e. genes not associated to this biological process
x <- 19 ## Number of "marked" elements in the selection, i.e. genes of the group of interest that are associated to this biological process
Compute the following statistics:
Let us first draw the distribution of probability densities, i.e. the probability to observe a particular value of "x": P(X=x).
## Define the range of possible values for x.
## The number of marked elements in the selection can neither be
## higher than the size of the selection, nor higher than the
## number of marked elements.
x.range <- 0:min(k,m)
help(dhyper) ## Always read the documentation of a function before using it.
## Compute the distribution of density P(X=x)
dens <- dhyper(x=x.range, m=m, n=n, k=k)
## Plot the distributon of hypergeometric densities
dev.new(width=7, height=5) ## Open a new graphical window
plot (x.range, dens, type="h", lwd=2, col="blue", main="Hypergeometric density", xlab="x = marked elements in the selection", ylab="density = P(X=x)", ylim=c(0, max(dens)*1.25))
## Draw an arrow indicating the expected value
arrows(exp.x, max(dens)*1.15, exp.x, max(dens)*1.05, lwd=2, col='darkgreen', angle=30, length=0.1, code=2)
text(exp.x, max(dens)*1.20, labels=paste("exp=", round(exp.x, digits=2)), col="darkgreen", font=2)
## Draw an arrow indicating the observed value
arrows(x, max(dens)*1.15, x, max(dens)*1.05, lwd=2, col='red', angle=30, length=0.1, code=2)
text(x, max(dens)*1.20, labels=paste("x=", x), col="red", font=2)
dev.copy2pdf(file=file.path(dir.results, "go_enrichment_hyper_density.pdf"))
With the parameters of our analysis, the hypergeometric density function has an asymmetric bell shape (in other cases, the distribution can be j-shaped or i-hsaped, or have a symmetrical bell shape). The red arrows indicates the number of E2F target genes which are annotated as "cell cycle". The hystogram already shows us an obvious fact: this number is much higher than what would be expected by chance (exp = 2.65), and it seems to have a very low probability.
A better way to emphasize small values is to plot the ordinates (Y axis) on a logarithmic scale.
## Plot the p-value distribution with logarithmic axes
plot (x.range, dhyper(x=x.range, m=m, n=n, k=k), type="l", lwd=2, col="blue", main="Hypergeometric density (log Y scale)", xlab="x = marked elements in the selection", ylab="density = P(X=x)", log="y", panel.first=grid())
## Arrow indicating expected value
arrows(exp.x, max(dens)*1e-10, exp.x, max(dens)*1e-2, lwd=2, col='darkgreen', angle=30, length=0.1, code=2)
text(exp.x, max(dens)*1e-15, labels=paste("exp=", round(exp.x, digits=2)), col="darkgreen", font=2)
## Arrow indicating observed value
arrows(x, dens[x+1]*1e-10, x, dens[x+1]*1e-2, lwd=2, col='red', angle=30, length=0.1, code=2)
text(x, dens[x+1]*1e-15, labels=paste("x=", x), col="red", font=2)
dev.copy2pdf(file=file.path(dir.results, "go_enrichment_density_logy.pdf"))
This logarithmic plot shows that the probability decreases very rapidly when the number of marked elements exceeds the expectation. In particular, the probability to observe by chance a selection made of marked elements only is very low (2e-81):
print(dhyper(x=k, m=m, n=n, k=k))
The P-value is the probability to observe at least "x" marked balls in the selection.
Compute the P-value for the observed number of marked elements in the selection
p.value <- phyper(q=x -1, m=m, n=n, k=k, lower.tail=FALSE)
We suggest you to read the documentation to check that the reasoning above is correct.
help(phyper)
## Compute the distribution of P-values (for all possible values of x).
p.values <- phyper(q=x.range -1, m=m, n=n, k=k, lower.tail=FALSE)
## Plot the P-value distribution on linear scales
dev.new(width=7, height=5) ## Open a new graphical window
plot(x.range, p.values, type="l", lwd=2, col="violet", main="Hypergeometric P-value", xlab="x = marked elements in the selection", ylab="P-value = P(X>=x)", ylim=c(0, 1), panel.first=grid())
## Arrow indicating observed value
arrows(x, max(dens)*1.35, x, max(dens)*1.1, lwd=2, col='red', angle=30, length=0.1, code=2)
text(x, max(dens)*1.5, labels=paste("x=", x, "; p-val=", signif(digits=1, p.value), sep=""), col="red", font=2)
## We can plot the density below the P-value
lines(x.range, dens, type="h", col="blue", lwd=2)
legend("topright", legend=c("P-value", "density"), lwd=2, col=c("violet", "blue"), bg="white", bty="o")
dev.copy2pdf(file=file.path(dir.results, "go_enrichment_hyper_pval.pdf"))
To better emphasize small P-values, we will display the P-value distribution with a logarithmic ordinate.
## Plot the P-value distribution with a logarithmic Y scale
dev.new(width=7, height=5) ## Open a new graphical window
plot(x.range, p.values, type="l", lwd=2, col="violet", main="Hypergeometric P-value (log Y scale)", xlab="x = marked elements in the selection", ylab="P-value = P(X>=x); log scale", panel.first=grid(), log='y')
## Arrow indicating observed value
arrows(x, p.value, x, min(dens), lwd=2, col='red', angle=30, length=0.1, code=2)
text(x, min(dens), labels=paste("x=", x, sep=""), col="red", font=2, pos=4)
## Arrow indicating the P-value
arrows(x, p.value, 0, p.value, lwd=2, col='red', angle=30, length=0.1, code=2)
text(0, p.value*1e-5, labels=paste("p-val=", signif(digits=2, p.value), sep=""), col="red", font=2, pos=4)
dev.copy2pdf(file=file.path(dir.results, "go_enrichment_hyper_pval_logy.pdf"))
Did we obtain the same P-value as DAVID ? If not, read David help page and try to understand the reason for the difference.
This will be further discussed during the practicals.
## Prepare a two-dimensional contingency table
contingency.table <- data.frame(matrix(nrow=2, ncol=2))
rownames(contingency.table) <- c("predicted.target", "non.predicted")
colnames(contingency.table) <- c("class.member", "non.member")
## Assign the values one by one to make sure we put them in the right
## place (this is not necessary, we could enter the 4 values in a
## single instruction).
contingency.table["predicted.target", "class.member"] <- x ## Number of marked genes in the selection
contingency.table["predicted.target", "non.member"] <- k - x ## Number of non-marked genes in the selection
contingency.table["non.predicted", "class.member"] <- m - x ## Number of marked genes outside of the selection
contingency.table["non.predicted", "non.member"] <- n - (k - x) ## Number of non-marked genes in the selection
print(contingency.table)
## Print marginal sums
(contingency.row.sum <- apply(contingency.table, 1, sum))
(contingency.col.sum <- apply(contingency.table, 2, sum))
## Create a contingency table with marginal sums
contingency.table.margins <- cbind(contingency.table, contingency.row.sum)
contingency.table.margins <- rbind(contingency.table.margins, apply(contingency.table.margins, 2, sum))
names(contingency.table.margins) <- c(names(contingency.table), "total")
rownames(contingency.table.margins) <- c(rownames(contingency.table), "total")
print(contingency.table.margins)
## Check the total
print(sum(contingency.table)) ## The value shoudl equal N, since every
## possible gene must be assigned to one
## cell of the contingency table.
print(N)
## Run Fisher's exact test
ftest.result <- fisher.test(x=contingency.table, alternative="greater")
print(ftest.result)
attributes(ftest.result) ## Display the list of attribute of the object returned by ftest
print (ftest.result$p.value) ## Print the P-value of the exact test
################################################################
## Compute expected values in the contingency table
exp.contingency.table <- contingency.table ## Quick and dirty way to obtain the same structure as the original contingency table
exp.contingency.table[ ] <- contingency.row.sum %*% t(contingency.col.sum) / N
## Add row and column sums (marginal sums)
exp.contingency.table <- cbind(exp.contingency.table, contingency.row.sum)
exp.contingency.table <- rbind(exp.contingency.table, apply(exp.contingency.table, 2, sum))
names(exp.contingency.table) <- c(names(contingency.table), "total")
rownames(exp.contingency.table) <- c(rownames(contingency.table), "total")
print(exp.contingency.table)
View solution|
Hide solution